





24小时服务热线:
400-1846-454


医疗器械与敷料微生物检测项目
医疗器械与医用敷料直接接触人体组织、黏膜或伤口,其微生物安全性直接关系到患者健康与治疗效果。一旦产品被微生物污染,可能导致严重的感染、败血症甚至危及生命。因此,全球监管机构(如美国FDA、欧盟MDR、ISO标准)对医疗器械的微生物控制提出了严格要求。
深圳中为检验化学实验室依据国际标准(ISO 11737系列、USP、EP等),提供全面的微生物检测服务,确保医疗器械与敷料符合GMP、注册申报及临床使用安全要求。
1. 生物负载测试(Bioburden Testing)
评估产品在灭菌前的微生物污染水平,是制定和验证灭菌工艺的关键前提。
检测内容:
需氧菌总数(TAMC)
霉菌和酵母计数(TYMC)
适用对象:所有非无菌医疗器械及其包装材料,如纱布、棉球、导管、手术器械、植入物等
标准依据:ISO 11737-1:2018《医疗器械的灭菌 – 微生物学方法 – 第1部分:产品上微生物群落的测定》
意义:为环氧乙烷(EO)、辐照(Gamma/X-ray)或蒸汽灭菌工艺参数设定提供科学依据
2. 无菌测试(Sterility Testing)
确认灭菌后的产品是否达到“无菌保证水平”(SAL ≤ 10⁻⁶),即每百万件产品中污染概率不超过1件。
测试方法:
直接接种法:适用于液体或可溶解产品
薄膜过滤法(推荐):适用于固体、半固体或难溶产品,如敷料、导管、植入物
培养条件:
有氧菌培养(TSB,14天)
厌氧菌培养(FTM,14天)
标准依据:
ISO 11737-2:2019《医疗器械的灭菌 – 微生物学方法 – 第2部分:无菌试验》
USP <71> Sterility Tests
EP 2.6.1
抽样要求:根据ISO 13408或AAMI ST72进行批次抽样
3. 细菌内毒素检测(Bacterial Endotoxins Test, BET)
检测革兰氏阴性菌细胞壁释放的脂多糖(LPS),防止引发发热、休克等热原反应。
方法:鲎试剂法(LAL test),包括凝胶法、显色法(Chromogenic)和动态浊度法(Kinetic Turbidimetric)
限值要求:根据产品与人体接触类型(如血液接触、组织接触)按USP <85> 或 EP 2.6.14 计算允许内毒素限值(K/M)
适用产品:
注射类器械(如注射器、输液器)
植入式器械(如人工关节、心脏瓣膜)
伤口敷料、透析设备等
标准依据:USP <85>、EP 2.6.14、ISO 29141
4. 灭菌验证支持检测
为灭菌过程(EO、辐照、湿热)提供微生物学数据支持。
D-value 测定:评估特定微生物对灭菌因子的抗性
生物指示剂(BI)性能测试:验证灭菌过程中使用的枯草杆菌芽孢(Geobacillus stearothermophilus)或嗜热脂肪芽孢杆菌(Bacillus subtilis)是否被有效杀灭
标准依据:ISO 11135(EO)、ISO 11137(辐照)、ISO 17665(湿热)
5. 环境微生物监测(Environmental Monitoring)
对生产环境(洁净车间、无菌灌装线)进行动态监控,确保生产过程受控。
检测项目:
空气浮游菌、沉降菌
表面微生物(设备、工作台)
操作人员手套/口罩微生物脱落
标准依据:ISO 14698、EU GMP Annex 1
6. 欧美市场法规要求概览
市场 | 主要法规 | 关键要求 |
美国 | FDA 21 CFR Part 820(QSR)、ISO 11737系列 | 所有III类及部分II类器械需提交生物负载、无菌和内毒素数据用于510(k)或PMA申报 |
欧盟 | 医疗器械法规(MDR EU 2017/745) | 要求制造商建立微生物控制体系,提供完整的灭菌与无菌验证报告 |
加拿大 | Health Canada Medical Devices Regulations | 参照ISO标准,要求高风险器械提供第三方检测报告 |
中国 | NMPA《医疗器械生产质量管理规范》 | 强调无菌检测与环境监控 |
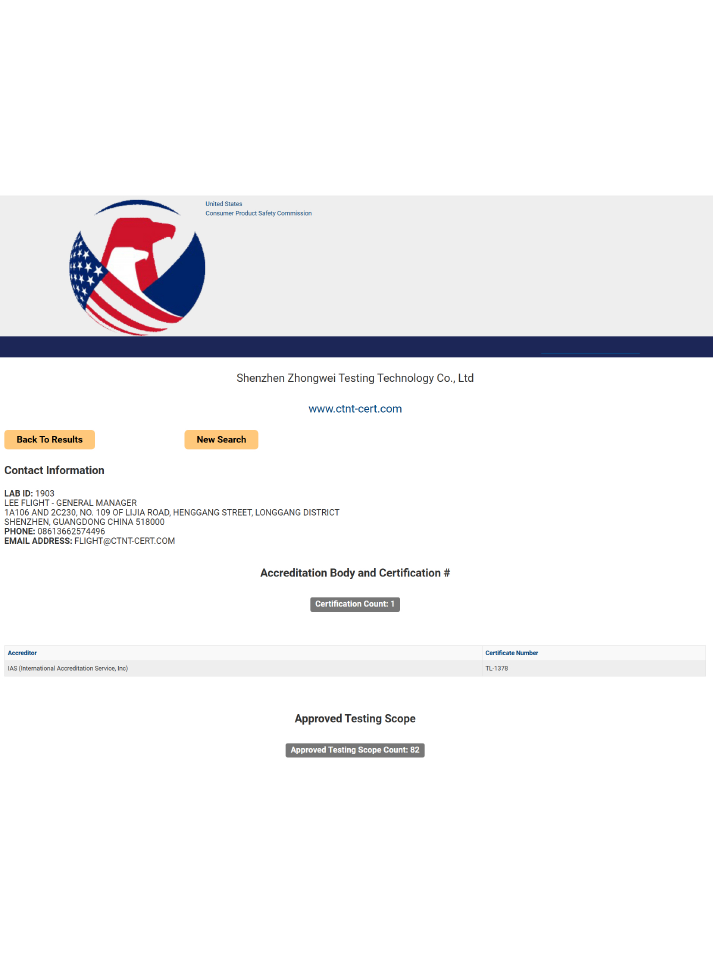
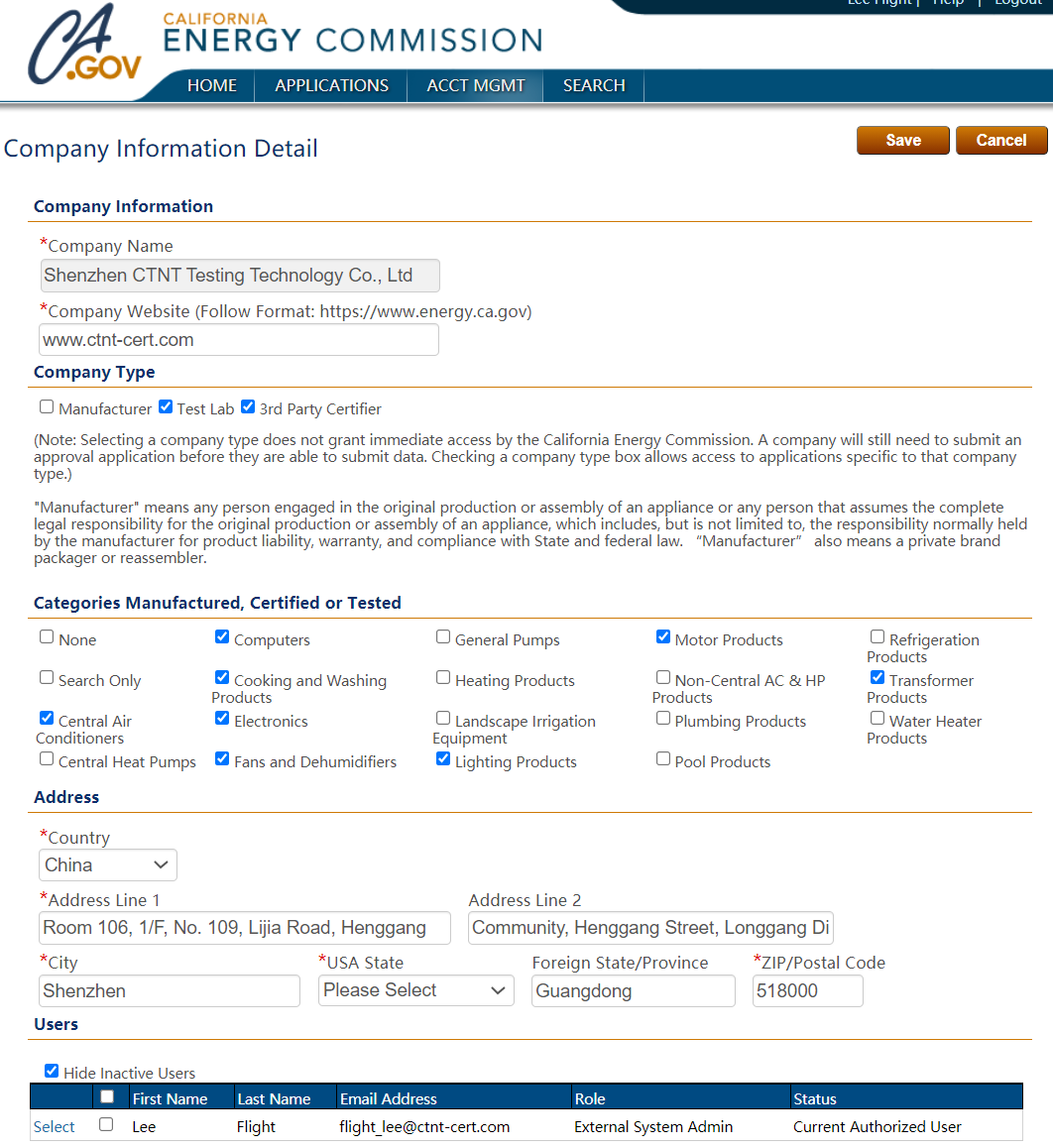
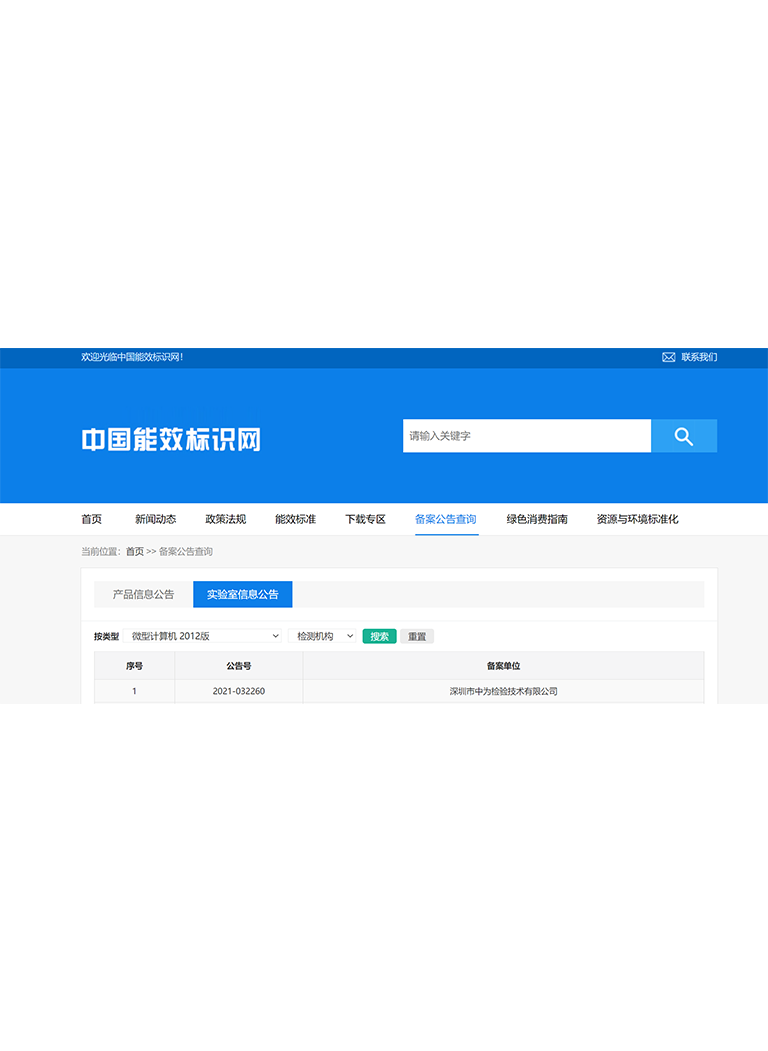
7.深圳中为检验微生物测试服务
深圳中为检验化学实验室可为各类医疗器械与医用敷料提供从原料到成品的全流程微生物检测服务,涵盖生物负载、无菌测试、内毒素检测及灭菌验证支持,严格按照ISO、USP、EP等国际标准执行,助力企业完成产品注册、CE认证及全球市场准入,保障医疗安全与患者福祉。
欢迎联系我们的专业客服获取相关服务!







电话:400-1846-454

地址:深圳市龙岗区横岗街道横岗社区力嘉路109号





 网站备案:
网站备案: 公安网备案:
公安网备案:

