





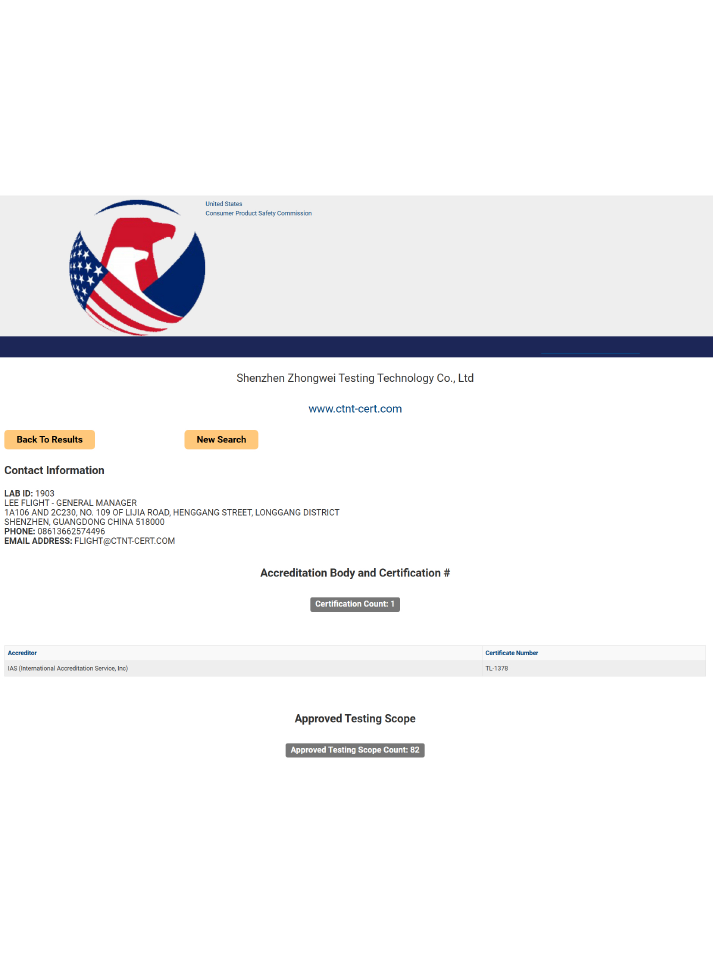
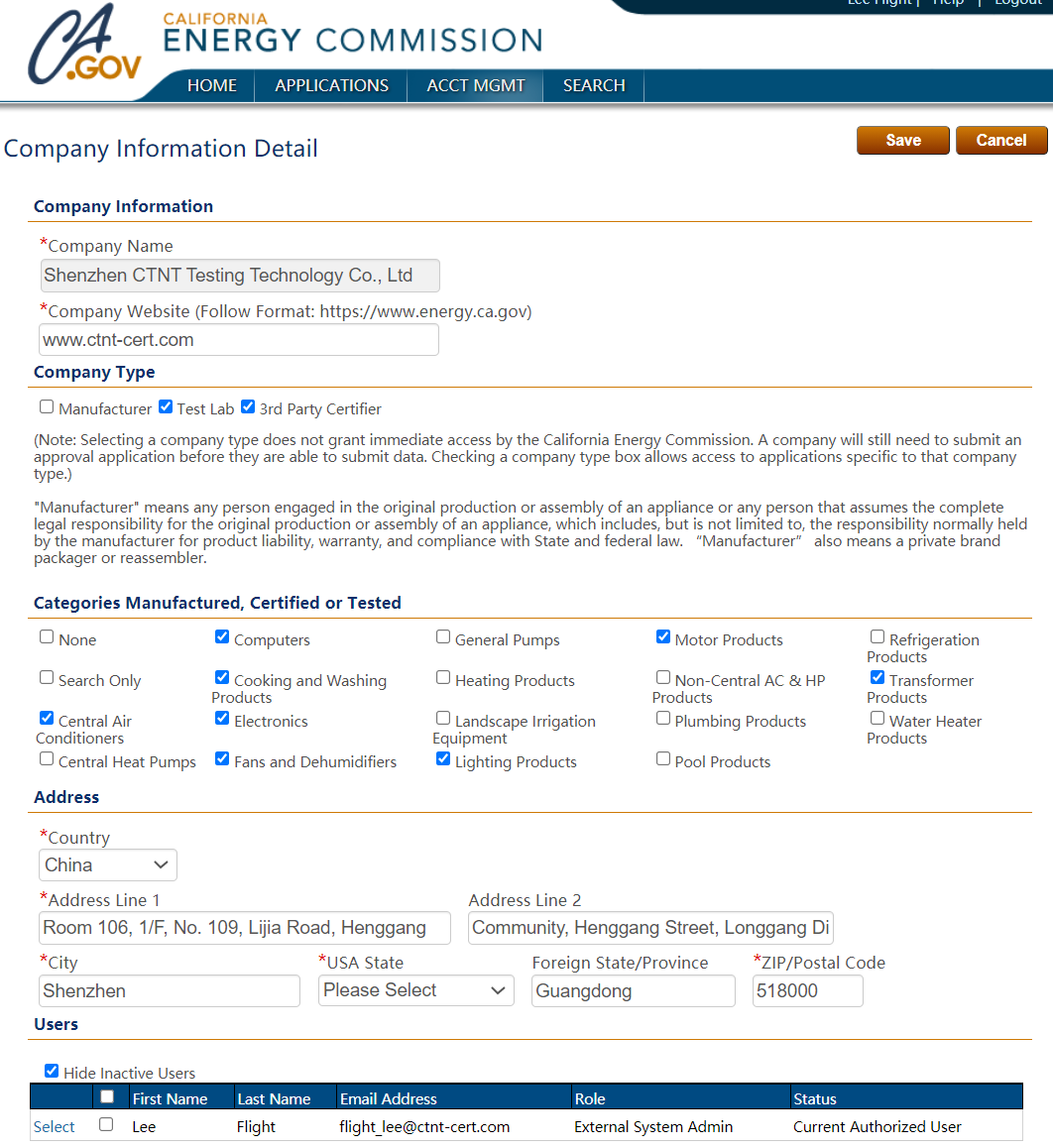
24小时服务热线:
400-1846-454




一、法规依据
FDA依据21 CFR Part 1040.10和1040.11对激光产品实施强制性监管。所有在美国销售、进口或分销的激光舞台灯
都必须完成注册和产品报告,否则将面临海关扣留、市场禁售等风险。
二、产品分类
激光舞台灯通常属于Class IIIb(500mW-1W)或Class IV(>1W)高功率等级,具有较高辐射能量,需严格管控。
分类依据包括输出功率、波长、脉冲特性等参数。
三、核心合规要求
安全设计要求
必须配备钥匙开关、紧急停止按钮、发射指示灯等安全联锁装置
扫描系统需内置失效保护:振镜故障时0.3秒内自动切断或降低激光输出
对于Class IIIb/IV产品,需配备光束遮挡装置和远程控制接口
标签标识要求
产品本体须标注激光等级(如"CLASS IV LASER PRODUCT")
必须包含波长、最大输出功率、制造商信息
使用国际通用激光危险符号(菱形图标加辐射线条)
警告语句示例:"DANGER: Laser Radiation. Avoid direct exposure to the beam."
用户手册要求
提供英文版使用说明书
包含安全操作指南、维护说明及潜在风险提示
明确警示使用场景限制(如避免直射人眼)
四、注册流程
前期准备
收集产品技术资料,包括电路图、光学结构图、BOM清单、用户手册等。
实验室测试
委托具备资质的第三方检测机构进行激光辐射安全测试,核心测试项目包括:
输出功率/能量测试(连续波或脉冲激光)
波长测定(主波长及次级波长)
光束发散角与直径测量
可达发射极限(AEL)验证
安全联锁功能测试
辐射泄漏测试
提交报告
通过FDA电子提交门户(eSubmitter或CDRH系统)上传激光产品报告(Laser Product Report),内容包括:
制造商信息、产品型号和描述
详细技术参数(波长、功率、光路结构)
测试数据和安全措施说明
符合性声明
五、企业注册与列名
制造商或美国进口商需在FDA完成企业注册(Establishment Registration),并在产品上市前进行产品列名
(Product Listing)。非美国本土企业必须指定一名美国代理人(US Agent)负责与FDA沟通。
六、年度报告与持续合规
FDA注册需每年更新。制造商需建立符合21 CFR 820的质量管理体系,保留完整测试记录至少5年以备FDA抽查。
七、违规风险
未合规产品可能面临:
FDA进口扣留(Import Detention)
产品召回(Recall)
罚款(单次违规可达数万美元)
制造商/进口商被列入黑名单,限制后续产品进入美国市场
关键提示:FDA对激光产品的监管是"注册与合规报告"制度,而非传统意义上的"认证"或"批准"。FDA不颁发纸质证书,
但会发放Accession Number作为合规凭证。许多电商平台和大型零售商要求供应商提供此号码作为上架凭证。






返回顶部

电话:400-1846-454

地址:深圳市龙岗区横岗街道横岗社区力嘉路109号
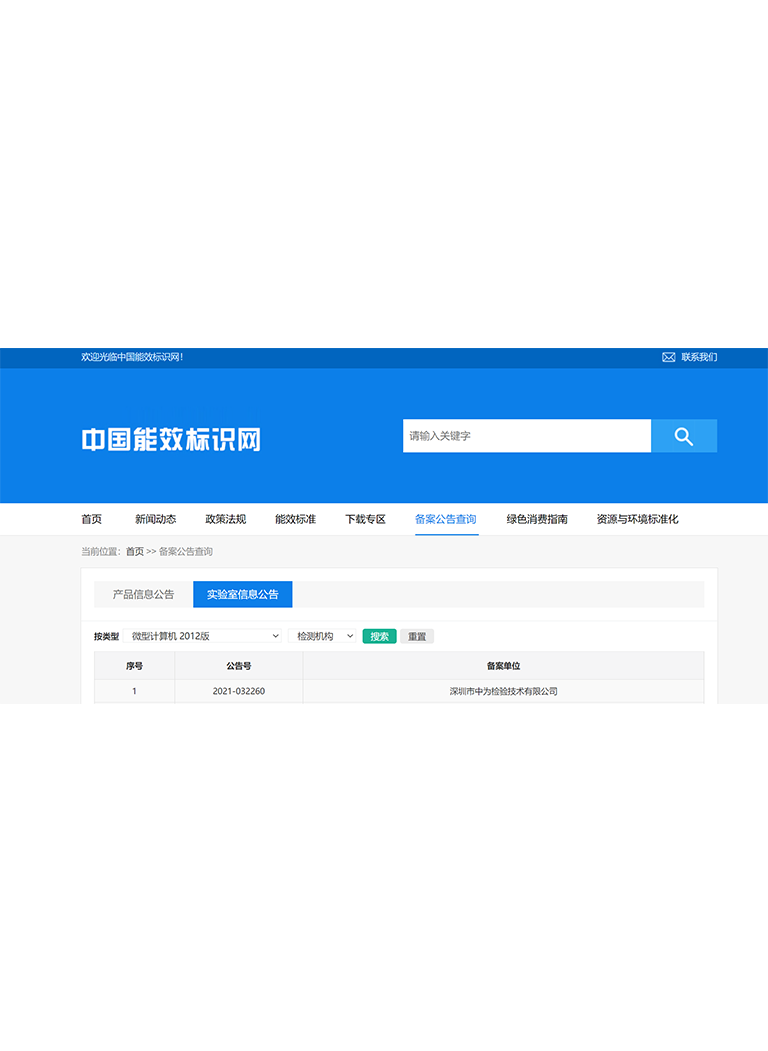
 当前位置:
当前位置: 





 网站备案:
网站备案: 公安网备案:
公安网备案:

